Nearly all mammalian cells contain cilia – microscopic hair-like structures that extend from the surface of cells. They can be up to 10 micrometers in length and up to 1 micrometer wide. Motile cilia can be found in the respiratory tract as well as the middle ear. These moving cilia work to keep these areas free of mucous accumulation. They are also known to help propel sperm within the reproductive tract. Non-motile cilia play a crucial role in many organs acting as sensory receptors for the cell by receiving signals from other cells or fluids nearby.
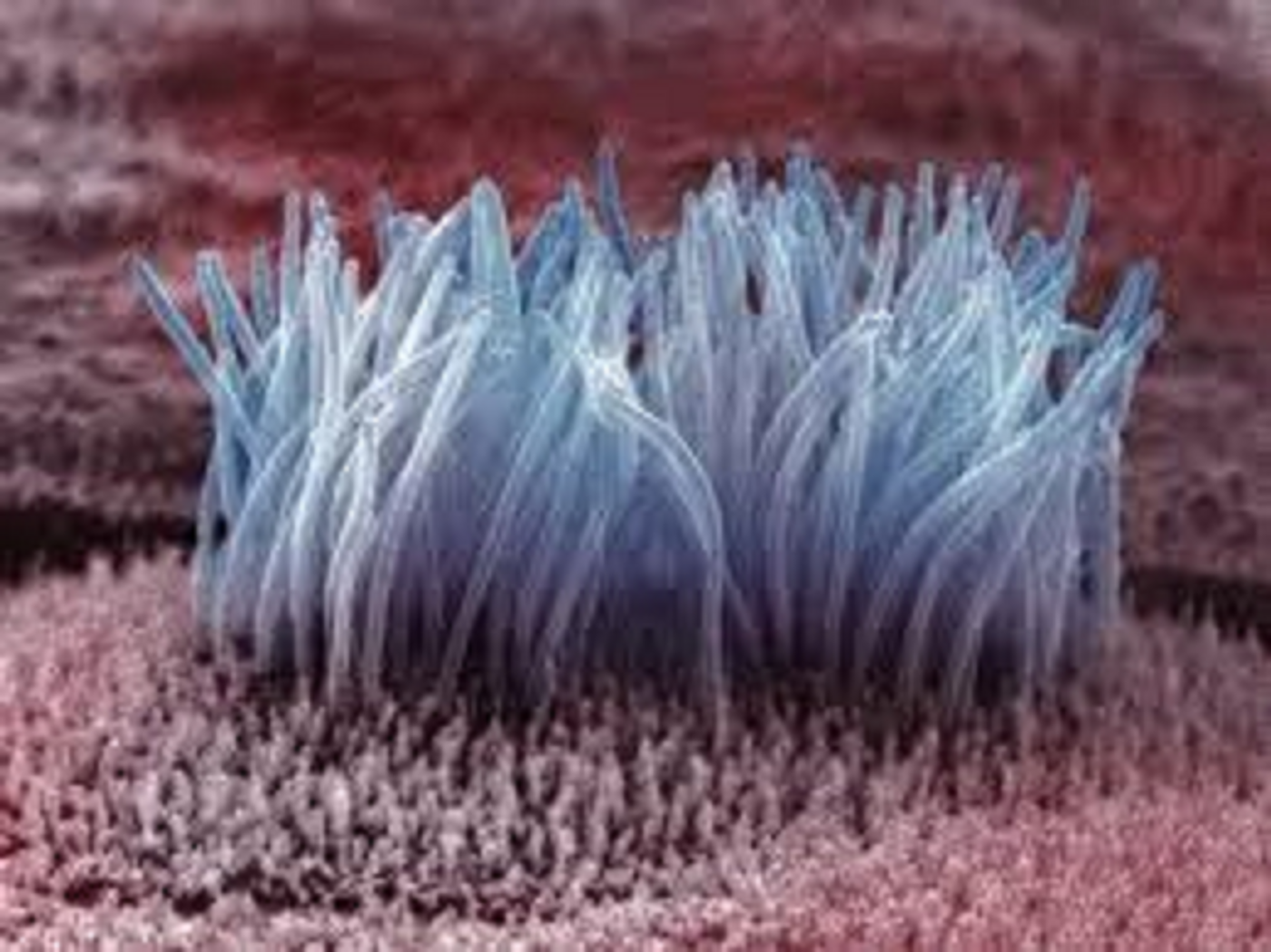
Ciliary defects cause a variety of symptoms including disease of the kidneys, blindness and nervous system defects. Cilia have sub compartments characterized by distinct molecular compositions. These molecular compositions encode for receptors and signaling components. There are also “gated” diffusion barriers at the base of cilia which regulate entry of proteins and other types of molecules into the cell. This ciliary gate is associated with an axon called the transition zone (TZ) which connects cell microtubules to the cell membrane. The TZ is thought to contribute to diffusion barrier properties of the cilia sub compartment.
Mutations in genes associated with the TZ are known to be the cause of ciliopathies such as Joubert Syndrome which is a rare developmental disorder affecting the brain and kidneys. In babies diagnosed with Joubert syndrome, developmental delay can cause breathing abnormalities and movement disorders. Retinal degeneration has been observed in some patients in combination with kidney disease. Newborns may also have extra fingers or toes (polydactyly) in some cases. It can be inherited in an autosomal recessive manner however; sporadic cases have also occurred.
Researchers from the University of Dublin sought to further dissect the molecular composition and mechanisms involving organization and barrier regulation of the TZ. They investigated the coexpression and coevolution of the protein TMEM107, which is mutated in persons with Joubert syndrome experiencing oral-facial-digital syndrome.
Using an animal model, they found that TMEM107 controls the composition of cilia and functions with another protein, NPHP4 to regulate the integrity of cilia as well as TZ docking. They also found that ciliopathy proteins are anchored at the TZ membrane which is crucial for proper function of the diffusion membrane. MKS2 proteins were found to be anchored in the TZ, playing an important role in barrier functions. This research provides insight into the architecture of the TZ domain as well as proteins involved in diffusion barrier formation, leading to ciliopathic diseases. Researchers suggest using similar genetic methods to characterize other ciliary models such as the IFT-A and IFT-B models.
Sources:
Nature Cell Biology;
National Institutes of Health;
Ciliopathy Alliance