What is bioinformatics?
The discipline of bioinformatics integrates biology and computer science to improve our understanding of complex biological systems. Bioinformatic analysis describes the processing of biological data from DNA, RNA, proteins, and other biological molecules using computational tools.
Where does bioinformatics fit into NGS workflows?
Bioinformatics plays many key roles in NGS workflows, starting at the very beginning (Figure 1). During project planning, insights from a bioinformatician can help researchers decide how many replicates (biological and/or technical) they need for each sample type, and also the required sequencing depth for a particular analysis. This is to ensure that robust analysis is possible, including the determination of statistically significant differences between samples.
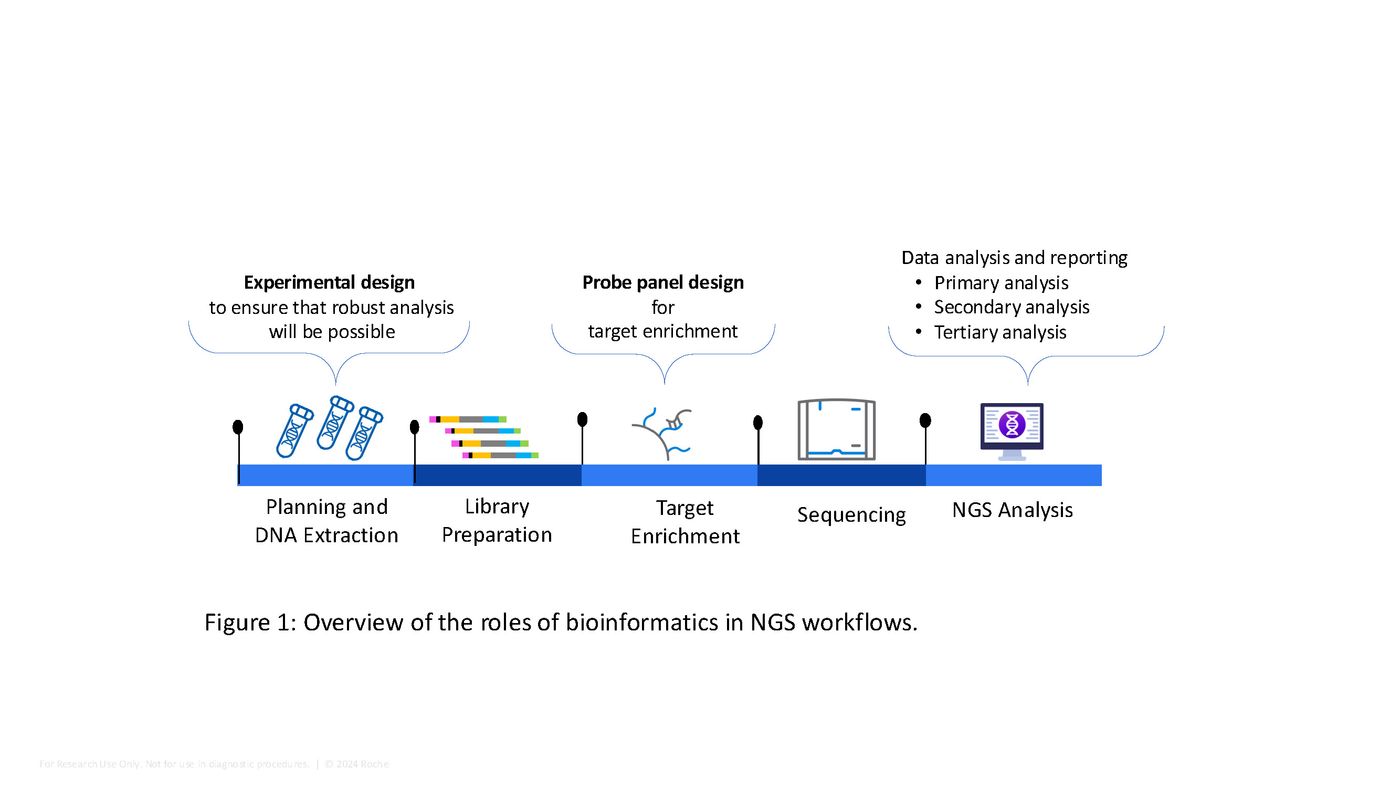 Many NGS workflows include target enrichment—the use of a probe or primer panel to selectively capture genomic regions of interest—to focus sequencing reads on specific areas. The creation of these panels requires computer algorithms; this is true whether an expert designer helps to create the panel, or whether the researcher uses an online panel design tool designed for non-bioinformaticians, in which case the algorithms operate “behind the scenes.
Many NGS workflows include target enrichment—the use of a probe or primer panel to selectively capture genomic regions of interest—to focus sequencing reads on specific areas. The creation of these panels requires computer algorithms; this is true whether an expert designer helps to create the panel, or whether the researcher uses an online panel design tool designed for non-bioinformaticians, in which case the algorithms operate “behind the scenes.
Finally—and this is where the bulk of the “bioinformatics magic” happens— bioinformatics experts and algorithms take the raw data that comes off the sequencer (millions of sequencing reads) and convert it into meaningful biological information, including statistical analysis where relevant. For DNA sequencing, these results can include uniformity of coverage, the presence and frequency of genetic variants, on-target rates, or the de novo assembly of genomes. For RNA sequencing, these results can include differences in gene expression, the relative activity of different pathways, and the detection of RNA fusions and splice variants.
What are the three main stages of NGS data analysis?
- Primary analysis begins with the conversion of the raw sequencing data, which comes off the sequencer as binary format data files (BCL files) into FASTQ files, which are text-based sequence files that include both the sequence data and the read quality scores. This step also includes demultiplexing, which is the process of sorting sequencing reads by individual sample indexes in cases where multiple samples are pooled (multiplexed). These steps are often carried out by software on the sequencing instrument.
- Secondary analysis begins with additional QC steps to assess the overall data. If the data passes QC, next steps include adapter trimming and quality filtering. This stage often includes the downsampling of datasets to contain the same number of reads; this enables data normalization, which is important when comparing variant frequencies, transcript frequency, or coverage depth across samples. Next, reads are mapped (aligned) to the genome being studied and duplicate reads are removed, followed by variant calling. Variants can also be viewed in Integrative Genomics Viewer (IGV).
- For target enriched sequencing, additional steps include target coverage analysis (depth, uniformity, and on-target rate). In many analysis pipelines, these steps involve numerous algorithms that are gathered together in a single collection called a “bioinformatics container solution” (see below).
For RNA sequencing, relative levels of gene expression are identified (often shown as a heat maps to make it easier to see patterns). This can be followed by pathway analysis (by examining which genes are most highly and lowly expressed in a given sample) and the identification of alternate transcripts such as RNA fusions and splice variants.
Tertiary analysis includes interpretation steps that describe the biological relevance of the NGS data. For example, this could include the identification of disease-associated variants or mutations that suggest a susceptibility to cancer or neurological conditions, or abnormally high or low expression of disease-associated genes. This stage can also include variant annotation in curated online databases.
What are “container solutions” in NGS bioinformatics?
A “bioinformatics container” refers to a group of software components that are linked together in a specific sequence within an analysis pipeline. This “containerization” of multiple algorithms enables scalable, reproducible analysis and documentation, and can increase analysis efficiency by automating the movement of data through each stage. This also can simplify the maintenance and sharing of complex bioinformatics resources, and can facilitate the alignment of results across research groups.
What roles can AI play in NGS bioinformatics workflows?
The implementation of artificial intelligence (AI) can further enhance and streamline the analysis of NGS data, especially as AI can automate and optimize numerous aspects of the process.
Want to learn more? For a discussion of how bioinformatics—including container solutions—can be used in the analysis of data from whole-exome sequencing (WES), watch the recorded webinar on Labroots linked here.

For Research Use Only. Not for use in diagnostic procedures.