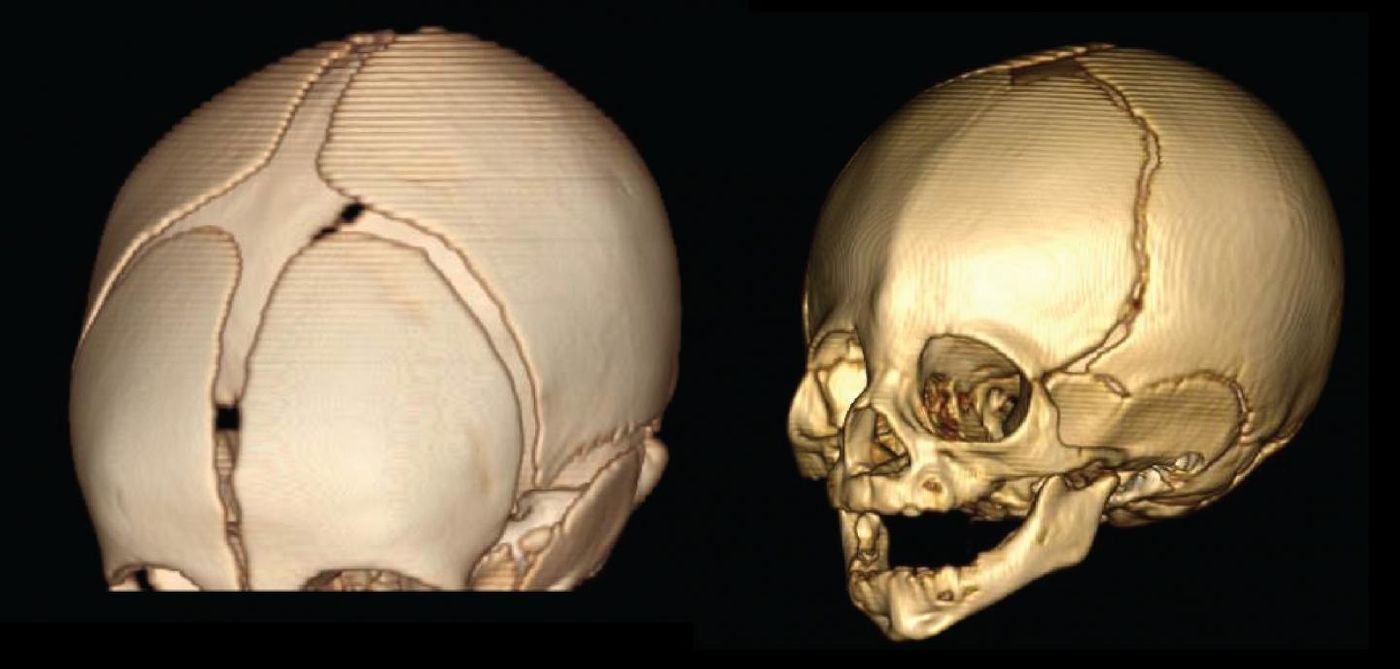
When the bones of an infant’s skull fuse too early, the growing brain is dangerously confined to a small space, causing various deformities of the facial and skull bones in addition to cognitive malfunction. In a new study from Rockefeller University, scientists have identified two rare mutations that cause the disease, called craniosynostosis, and the unique series of events that precede them.
Nearly 200 families were recruited to participate in the study, which focused on identifying mutations causing a specific type of craniosynostosis, called midline or sagittal craniosynostosis. Signs of the disease occur quickly after birth, with an oblong shape of the head being a major indicator.
Researchers conducted multiple genetic tests to investigate the pattern of inheritance for disease-causing gene mutations. After sequencing the participants’ exomes, the part of the genome that contains the protein-coding genes, they connected mutations in a gene called SMAD6 to children with craniosynostosis. However, none of the parents of children with the disease shared the condition, even if they also had the SMAD6 mutation.
SMAD6 inhibits BMP signaling, a processes that promotes bone formation. The SMAD6 mutation in the children with craniosynostosis prevented proper inhibition of bone formation in the skull, leading to early skull fusion. However, since other participants with the same SMAD6 mutation were perfectly healthy, the researchers knew there had to be another part of the genetic equation that they were missing.
The next suspect was a gene called BMP2, which is connected to the same bone formation pathway as SMAD6. They found that the same children from the study who had the SMAD6 mutation and craniosynostosis also had a mutation in BMP2. Without SMAD6, these children were already missing a key piece of the puzzle for bone formation. The BMP2 mutation only further worsened the condition of premature bone formation and fusion.
It appears that both BMP2 and SMAD6 mutations are needed to cause midline craniosynostosis. Confirming the relationship will help families and doctors decide the risk of disease.
“In each case, the SMAD6 mutation came from one parent and the BMP2 risk variant came from the other parent, explaining why neither parent had craniosynostosis," said first author of the study, Andrew Timberlake.
Scientists believe their research might also apply to other rare genetic diseases, potentially answering lead author Richard P. Lifton’s question: “Why do some individuals with potent rare mutations develop disease, while others with the same mutations do not?"
Lifton’s study was recently published in the journal
eLife.
Sources:
Rockefeller University,
Johns Hopkins Medicine